PC MODELING
The basis of molecular mechanics is that the energy of a molecule can be described in terms of a function called the force field that depends only on nuclear motions. Electrons are not considered explicitly. According to the Born-Oppenheimer approximation of the Schrödinger equation, nuclei are much heavier and move much more slowly than electrons, which attain their optimum distribution fast enough to adjust to any movement of the nuclei. The force field must provide a good description of the forces acting within and between the molecules while keeping the calculations within the limits of current computational resources. There are many different force fields which use different forms for the various interactions based on a set of parameters obtained either from experimental or theoretical data. Those parameters are assumed to be the same for different molecules, for example C-H stretching frequencies and bond lengths are nearly independent of molecular environment. Thus we would expect the force field to treat C-H bonds equally whether the Hydrogen atoms are bonded to an Acetaminophen or a methane molecule.
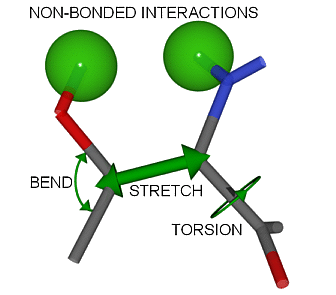
A force field is a collection of atom types, equations and parameters. Carbon atoms, for example can be sp, sp2 or sp3-hybridized having linear, trigonal or tetrahedral bonding geometry respectively. The force field contains parameters for these different types of bonds. The energy and geometry of a molecule is evaluated by potential energy equations associated with bond stretching, bond bending, torsional strain , non-bonded interactions.
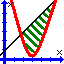
The stretching energy equation is based on Hooke's law.
where kb is the stiffness of the bond (modeled as a spring) and R0 is the equilibrium length.
The bending energy equation is also based on Hooke's law.
where kθ is the stiffness of the angle and θ0 is the equilibrium value.
The torsional energy represents the amount of energy that must be added to or subtracted from the sum of all other energy terms to make the total energy agree with experiment or quantum mechanical calculations,
This is a periodic function where the A parameter controls the amplitude, n controls the periodicity, and Φ shifts the entire curve along the rotation angle axis (τ).
The non-bonded energy represents the pair-wise sum of the energies of all possible interacting non-bonded atoms i and j,
The non-bonded energy accounts for repulsion, van der Waals attraction, and electrostatic interactions. Van der Waals attraction occurs at short range, repulsion occurs when the distance between interacting atoms becomes shorter than the sum of their contact radii (close to bonding distance). The A and B parameters control the attractive force hardness of the atoms respectively.
The electrostatic contribution is modeled by a Coulombic potential. The electrostatic energy is a function of the charge on the non-bonded atoms, their interatomic distance, and a molecular dielectric expression that accounts for the electrostatic interaction with the environment. Often, the molecular dielectric is set to a constant value between 1.0 and 5.0. A linearly varying distance-dependent dielectric is sometimes used to account for the increase in environmental bulk as the separation distance between interacting atoms increases. The sum of all terms gives the energy of the system of atoms,
GO TO 
![]() PROGRAMS
PROGRAMS
![]() CHEMISTRY
CHEMISTRY
![]() PC MODEL
PC MODEL
TO BUILD A STRUCTURE
You should draw your structure in small sections and then minimize it. It is difficult to fix problems for large structures.
The program starts in structure-drawing mode. Click on the [DRAW] command, then click in the drawing box. An atom will be drawn. By default, the first atom you draw is carbon. If you move the mouse within bonding distance and click again, a second atom is drawn and a bond is created between the atoms.
Instead of adding more atoms, a good way of building a stucture is by adding hydrogens to the first atom using the [H-A/D] command. This would place hydrogen atoms according to the oxydation state of the central atom. Then, to change atom type, click on an atom type in the periodic table [PT] menu and then click on the atom.
To close a ring, click an existing atom. To start drawing from an existing atom without adding a new bond to that atom, click on [DRAW] again, click on the old atom, and continue drawing. To start drawing from a new location, click on DRAW and click in a new area in the drawing box. To add fragments, click on selections in the fragment library. The atom template can be changed to a biomolecular or metal template. Click on [UPDATE] to stop drawing.
Alternatively, if the structure is too difficult to build, you can start from an existing structure. PC MODEL contains a database of amino acids, nucleotides and other templates. For example, a molecule of ATP can be built starting from a nucleotide, then adding atoms and changing atoms.
The program can also read some file formats that can be downloaded from the web. One of the most widely used formats is PDB, such files are available from our lab home page.
SUMMARY OF PC MODEL MENUS AND COMMANDS
Three different menus are available: [INPUT], [MINIM], and [DISPL]. To select a menu item or draw atoms, position the mouse cursor over the item and click the left mouse button. To select an item in a white pop-up menu, click the right mouse button. To rotate the object, position the mouse cursor in the drawing box and click & hold the MIDDLE mouse button and move the mouse.
In the INPUT, MINIM, and DISPL menus, the READ, WRITE, and STOP options are always displayed. PC MODEL can READ and WRITE the following formats:
PCM PCMODEL new file format
MMX MMX standard file format
MM2 MM2(77) file format
MNDO File format for molecular orbital program MNDO
MOPAC Z-matrix form for MOPAC and AMPAC
X-RAY generic x-ray file:
header, cell parameters, X Y Z coordinates
MACROMODEL MACROMODEL file format
SYBYL file format for SYBYL (Tripos, Inc)
C3D Chem3D format for Mac program Chem3D
PDB (READ only) Protein Data Bank format
ALCHEMY (WRITE only) Alchemy format
INPUT Menu
[DRAW]
Draws a carbon atom
[DELET]
click on atoms and bonds to delete, then click on [DONE]
[ADD-B]
click on a bond to increase the bond order by 1. To draw a new bond, use [DRAW]
[UPDAT]
updates the structure
[H-A/D]
first click on [H-A/D] deletes hydrogens not involved in hydrogen bonds, second click adds hydrogens according to a geometric perscription.
[MOV-A] moves atom within the plane of the screen. Click on atom, then click at new location. Click [UPDAT] to show the atom in the new location. Then click [H-A/D] to quickly move hydrogens attached to atoms that you move.
[M-IN]
click on atom to move atom 0.5 angstroms into the plane of the screen.
[M-OUT]
click on atom to move atom 0.5 angstroms out of the plane of the screen.
[ROT-S]
use middle mouse button instead of [ROT-S] to rotate the entire structure. If a substructure is active, use [ROT-S] to rotate the substructure, and select [FAST ROTATE]
[ERASE]
erases a structure. First use [WRITE] to save the structure. [ROT-B]
point to 4 atoms in dihedral angle---FIRST atom that you click on will move. Then click on slide bar (-45---0---+45) at top of draw box to smoothly rotate the angle. The farther you click on the edges of the slide bar, the faster you will rotate.
[SSADD]
reads previously stored structure file (in any format) into the program as a substructure
[MAKSS]
The substructure is created by pointing to an atom within an isolated structure, then to a blank spot in the drawing box. This will turn the entire isolated structure into a substructure. Or a portion of a structure can be identified as a substructure by first clicking on the "end" atom in the substructure followed by the the atom bonded to the "end" atom that is NOT in the substructure.
[MOVSS]
moves a substructure. Click on atom of substructure, then click on position to move atom. Entire substructure will then move.
[CONEC]
connects substructure to another structure. Both structures must have hydrogens. Click on a hydrogen in each structure; the hydrogens disappear and the bond will be made. The substructure will change orientation to make a reasonable bond.
[S-VIS]
sets a substructure to be invisible
[BNDLB]
changes the display to show bonds only, atom numbers, MMX atom types, substructure numbers and colors.
[REDUC]
shrinks size of structure
[EPIMR]
point to the center to be epimerized and then to the two attached atoms to be exchanged. Does not work if atoms are in a ring.
[XY,XZ,ZY]
view structure from XY, XZ, or YZ plane. Fast way to rotate the structure by 90
[RETYP]
click on hydrogens that should NOT be removed by H-A/D (i.e., are non-volatile)
[RESET]
reinitializes options such as fixed atoms, pi atoms, atomic charge, etc. to default values.
[H-BND]
marks acidic hydrogens with a "~". Minimizations will recognize hydrogen bonding between these "~" hydrogens and other atoms.
[PIATM]
automatically or manually mark atoms in pi systems. To remove piatm designation, select [PIATM] and click on the atom.
[COATM]
coordinates metal with lone pairs or pi systems, and input charge on metal. Default metal charge = 0; setting a charge is NOT optimized---be cautious when interpreting results. If you do NOT use [COATM], all metals are assumed saturated and NO attractive potential between metal and ligands.
[FXDIS]
prefered distances between atom pairs may be defined. Use a force constant 50 mdyne/A to keep atoms fixed.
[FXATM]
fixed atoms will not move during minimization.
[FXTOR]
fixed torsion angles will not move during minimization
[PRINT]
generates postscript file for printing.
[CLIP]
to zoom in on a region of the structure, point the mouse cursor in the center of the area to be expanded and click the left mouse button. To zoom out (make the structure smaller), click the right mouse button.
MINIM Menu
[MMX-M]
Performs a minimization. The atomic movement (in 10-5 A) and energy (kcal/mol) are listed every 5 iterations. the program will periodically pause to check the effect of each atom's movement on energy. Type
[MMX-E]
Computes energy, no minimization is performed
[ROT-E]
generates a graph of rotation energy barriers for acyclic torsion angle. The rotating parts are NOT minimized at each angle, but are held rigid. After displaying graph, you can return to original angle or new angle. A more accurate barrier may be obtained by rotating to a maximum, performing MMX-M, and repeating [ROT-E].
[BATCH]
allows unattended processing of multiple structures in background (not shown on screen)
[DOCK]
performs a simulated annealing to minimize the energy between rigid structures by varying the position and orientation of the structures.
[DYNAM]
runs molecular dynamics simulations.
[DCONT]
continues molecular dynamics simulations
[D-DRV]
generates a graph of rotation energy barriers for acyclic torsion angle. The rotating parts ARE minimized at each angle.
[DP-DP]
uses bond dipoles for polar interaction term in electrostatic calculations (the origional MM2 protocol). Default = atomic charges (which is especially important for systems with charged atoms, e.g., N+ or C-)
[BELOF] turns off the bell that rings when minimization is done
[NAME]
used to change the name of the structure in the program
[PFLAG]
sets level of detail of minimization info sent to the file pcmod.out
[S-MIN]
selects substructure to minimize
[CONST] used to add your own constants to the minimization routine
[ITERS]
used to alter the number of minimization iterations. Default = 30,000
[IT/S]
used to alter the number of iterations to complete before pausing to ask if the minimization should continue. Default = 500
[DIELC]
set dielectric constant. Default = 1.5. If negative, then the dielectricis also multiplied by the interatomic distance, creating a "distance dependent" dielectric
DISPL Menu
[AXY,AYZ,AXZ]
redraws structure with 3 selected atoms in the specified plane
[AX,AY,AZ]
redraws structure with 2 selected atoms parallel to specified axis
[BNDWD]
increases bond widths
[PAUSE]
pauses program. Designed for PCs, not helpful on SGIs.
[H-O/O]
turns on/off display of hydrogens (hydrogens are STILL there)
[R/S]
click on four atoms arond common center in order of priority, then click in blank area of screen. R or S designation is displayed at blank area.
[ENANT]
click on atom, structure is reflected through Z axis (perpendicular to screen) at that atom.
[QUERY]
click on 1, 2, 3, or 4 atoms, then click on blank area of screen. The XYZ coordinates, distance, angle, or torsion angle (respectively) will be displayed. Click on [UPDAT] to clear the [QUERY] display.
[PMR] click on two atoms, then at blank area of screen. Displays 1H NMR coupling constants between 1,4 bonded protons in H-C-C-H chain (neither C can be a methyl group).
[COMP]
reads a previously saved structure into program and superimposes new structure on existing structure.
[SURF]
draw dot, CPK, space-filling, electrostatic, 3D surface. Can also generate postscript file of these surface drawings.
[ORTEP]
draw ortep 3D surface. Can also generate postscript file of these surface drawings.
[EXPND]
display PCMODEL window and structure without menus. Used to photograph screen. Type
[DIMAP]
plots DIHED.XYZ (results from D-DRV in MINIM menu) or Weiler-type dihedral map of a ring
[PEPTD]
draws a ribbon through peptide backbone. Select "oxygen-oxygen" for smooth helical ribbons, choose "swapped orientation" for smooth sheet ribbons.
[STERO]
switches the display between stereo and mono
External Links
THIS PAGE IS

UNLESS THE BROWSER
HAS BUGS