

CHEMICAL KINETICS
Reaction rates are seldom related to thermodynamic functions of state of reactants and products. What determines the rate are the properties of the reactants and the intermediate transition state or activated complex. The standard data of chemical kinetics are the concentrations of reactants or products as a function of time. The method of choice for monitoring such quantities will depend mainly on the relative speed of the reaction and the nature of the reactants or products. For example; a reaction that consumes or generates gas can be monitored by the change in the volume of the gas component; if the reactant or product is a chromophore, its concentration can be monitored by the change in absorbance as a function of time.
One of the goals of chemical kinetics is to describe the rate at which the reactants are transformed into the products of the reaction. The term rate has several meanings, in chemical kinetics, the rate of a reaction is the change in the concentration of one of the reactants during a certain period of time. Take for example the reaction,
Then we write the concentrations of the reactants and products X, Ω, and Φ as [X], [Ω], and [Φ]. In general, the rate of formation of product can be written as the rate of reactant consuption,
The stoichiometry of the reaction can be more complex,
Then the rate becomes:
those expressions are very simple to derive no matter how complex the chemical equation might get.
The rates of reactions change with time; usually the rate of a reaction gradually decreases as the reactants are consumed. It is then advisable to measure reaction rates over short periods of time (dt); thus small changes in the concentration of a species (dXi). The ratio of these two quantities is called the instantaneous rate of reaction.
.
The instantaneous rate of reaction at any moment in time can be calculated from a graph of the concentration of the reactant (or product) versus time...
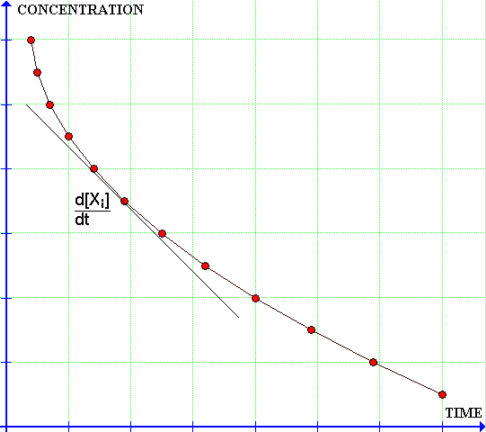
This is a typical graph showing a typical result: the rate of reaction at every point on this curve is directly proportional to the concentration at that point. By concentration we mean the concentration of a reactant involved in the rate limiting step of the reaction. Then the rate law would have the form:
This means that the reaction rate depends only on the concentration of reactant A. In reality, most reaction rates depend on the concentration of more than one reactant:
The rate law is an equation that relates the reaction rate to the concentrations of reactants or catalysts. Note that the rate law contains a constant k; a proportionality constant between rate and concentration. The rate constant has a fixed value at a given temperature. The superscripts a and b denote the reaction order; the exponent of the concentration of that species in the rate law as determined experimentally.
REACTION RATES FROM REACTION DATA
As an example, let's determine the rate law for the reaction:
where C is a catalyst. The following reaction data was obtained:
| Init [Φ2Ψ2] | Init [C] | Rate (time units) | |
| Trial 1 | 0.10 | 0.020 | 2300 |
| Trial 2 | 0.20 | 0.020 | 4600 |
| Trial 3 | 0.30 | 0.020 | 6900 |
| Trial 4 | 0.10 | 0.040 | 4600 |
| Trial 5: | 0.10 | 0.060 | 6900 |
-During the first three trials, the concentration of Φ2Ψ2 has been varied and the concentration of C has been kept constant.
-The concentration of Φ2Ψ2 in Trial 2 double of what it was in Trial 1, this leads to a two-fold increase in the reaction rate.
-The concentration of Φ2Ψ2 in Trial 3 three times as large as it was in Trial 1, this leads to a three-fold increase in the reaction rate.
Thus we conclude that the reaction rate is directly proportional to the Φ2Ψ2 concentration.
-Experiments 1, 4, and 5 were run at the same initial concentration of Φ2Ψ2 but different initial concentrations of the catalyst C.
-Comparing Trials 1 and 4 we see that doubling the initial C concentration leads to a two-fold increase in the reaction rate.
-Trials 1 and 5 show that tripling the initial C concentration leads to a three-fold increase in the reaction rate. Thus we conclude that the rate of the reaction is also directly proportional to the concentration of C.
The results are consistent with a rate law that is first-order in both Φ2Ψ2 and C.
Because the rate law is first order in both Φ2Ψ2 and C, the likely reaction mechanism is:
2Φ2Ψ2 + 2C --> 2CΨ + 2Φ2Ψ .....Rate determining step
2CΨ --> 2C + Ψ2
_________________________________________________________
2Φ2Ψ2 --> 2Φ2Ψ + Ψ2 .............Overall reaction
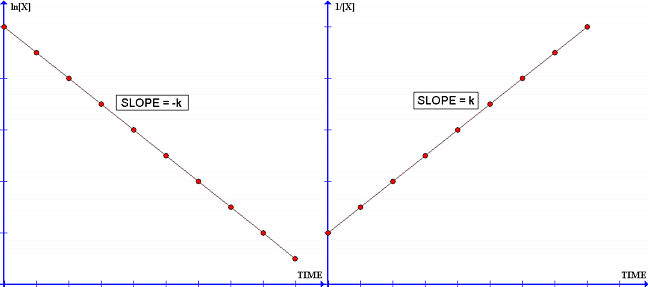
The order of the reaction can also be determined by plotting the data in several different ways until a straight line is obtained...
-Assume the reaction is first-order. Plot log[reactant] as a function of time,
-Assume the reaction is second-order. Plot 1 / [reactant] as a function of time,
-Assume the reaction is third-order. Plot 1 / [reactant]2 as a function of time, and so on.
THEORETICAL TREATMENT OF REACTION RATES
Consider the reaction,
Whose rate, assuming that d[Φ]/dt = -d[X]/dt = -d[Ω]/dt, can be defined as:
Here, the reaction is first order in both X and Ω, meaning this is a bimolecular reaction. If the concentration of reactants and products [X], [Ω], and [Φ] are written as number of particles per cubic centimeter N'X, N'Ω, and N'Φ, we can use Maxwell's distribution function to compute the number of collisions between the reactant particles as a function of time,
Where σ2 is the collision cross-section, μ is the reduced mass, [mXmΩ]/[mX + mΩ], and kT is Boltzmann constant times the absolute temperature. For the reaction to occur, the reacting particles must collide and if every collision resulted in reaction the rate would be equal to ZXΩ,
However, not all collisions result in reaction. Thus, kbi is actually equal to k'bi times the Boltzmann factor e-Ec/kT,
Where Ec is the critical value of the relative translational energy that two colliding particles must have in order to yield a reaction.
The above collision theory is based on the hard-sphere model of gasses, but can be applied to solution reactions if k' is multiplied by a probability factor,
Rate constants for most chemical reactions closely follow the Arrhenius equation,
Where A = Pk', is the frequency factor and can be assumed to be temperature-independent, e is the base of natural log, Ea is the activation energy, and R is the gas costant.
Taking logarithms:
Using logarithms of base 10:
This is the equation of a straight line if...
logk ---------------> y
logA ---------------> b
{-Ea / 2.303 X R} --> m
{1/T} --------------> x
Since we have obtained the energy of activation from the value of k at different temperatures, we should be able to obtain the value of of k at different temperatures from the activation energy by taking the difference of two equations at different temperatures,
logk2 = logA -{Ea / 2.303 X RT2}
-logk1 = logA -{Ea / 2.303 X RT1}
________________________________________________________
= logk2 - logk1 = -{Ea / 2.303 X RT2} + {Ea / 2.303 X RT1}
= log{k2 / k1} = {Ea / 2.303 X R} X {[1 / T1] - [1 / T2]}
This is the two-point Arrhenius equation (an easy way to get a straight line is to plot only two points :)
An accurate determination of the activation energy requires at least three runs completed at different reaction temperatures. The temperature intervals should be at least 5 0C. It has been found experimentally that many reactions exhibit Arrhenius-like dependence on temperature, thus a plot of ln[k] as a function of the reciprocal temperature in Kelvin gives a straight line. For Example, compound Γ decomposes into compound Ξ in the following reaction,
Since compound Ξ is a gas and compound Γ is a solid, the reaction was monitored by the change in the pressure of the system. The pressure readings were then converted to mol per unit time using the ideal gas law. The rate constants over the temperature range 100 to 300 K are reported in the table below.
| Temperature [K] | 100 | 150 | 200 | 250 | 300 |
| k [L mol-1 sec-1] | 5.00 | 15.0 | 30.0 | 50.0 | 75.0 |
We process the data by plotting ln[k] as a function of the reciprocal temperature in Kelvin
| 1/T [K] X 102 | 1.00 | 0.667 | 0.500 | 0.400 | 0.333 |
| lnk [L mol-1 sec-1] | 1.609 | 2.708 | 3.401 | 3.912 | 4.317 |
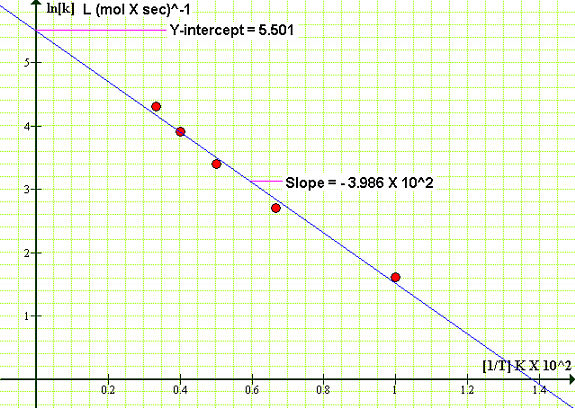
We note that the Y-intercept is 5.501 and the slope is -3.986 X 102, thus we obtain the Ea from,
-Ea = Slope X RAnd the frequency factor,
Ea = 3.986 X 102 K X 8.314 J K-1mol-1 = 3.313 X 103 J mol-1
A = e5.501 = 245 L mol-1 sec-1
The units might look complicated, just remember that the units must cancel to yield the answer you seek in the correct units.
Notice this data yields a straight line. If a straight line cannot be obtained, we can define the activation energy as
Ea = RT2[dlnk/dT]
REQUIRED MATERIALS AND APPARATUS
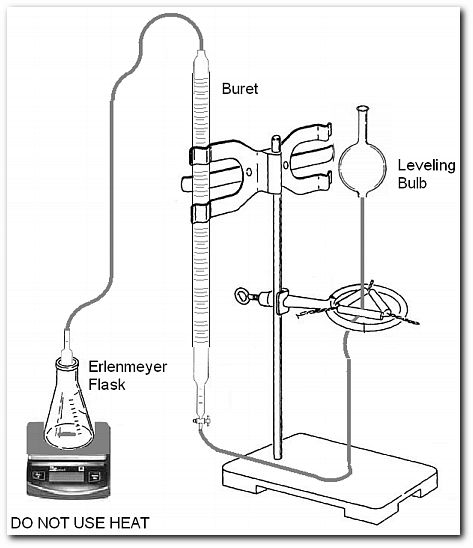
Setup for the chemical kinetics experiment.
EXPERIMENTAL PROCEDURE
Check the aparatus for leaks. Note the position of the meniscus before attaching the reaction flask. Allow gas to enter the buret and depress the liquid level. Periodically adjust the leveling bulb to maintain constant pressure.
Record the volume and time. Have all materials ready, you are doing a total of 15 runs. The amounts of each reactant and solvent are:
| T (0C) | H2O2 (mL) | KI (mL) | H2O (mL) | |
| Trial 1 | 25 | 7.5 | 2.0 | 15.5 |
| Trial 2 | 25 | 7.5 | 4.0 | 13.5 |
| Trial 3 | 25 | 7.5 | 6.0 | 11.5 |
| Trial 4 | 25 | 7.5 | 8.0 | 9.5 |
| Trial 5 | 25 | 7.5 | 10.0 | 7.5 |
| Trial 6 | 25 | 2.0 | 7.5 | 15.5 |
| Trial 7 | 25 | 4.0 | 7.5 | 13.5 |
| Trial 8 | 25 | 6.0 | 7.5 | 11.5 |
| Trial 9 | 25 | 8.0 | 7.5 | 9.5 |
| Trial 10 | 25 | 10.0 | 7.5 | 7.5 |
| Trial 11 | 30 | 7.5 | 7.5 | 10.0 |
| Trial 12 | 35 | 7.5 | 7.5 | 10.0 |
| Trial 13 | 40 | 7.5 | 7.5 | 10.0 |
| Trial 14 | 45 | 7.5 | 7.5 | 10.0 |
| Trial 15 | 50 | 7.5 | 7.5 | 10.0 |
DATA PROCESSING